

Cystic fibrosis (CF) is a rare disease that causes damage to the respiratory, digestive and reproductive systems. Its incidence varies across the globe. In the USA, CF affects one in every 3500 newborns, and in Europe it’s one in every 2000–3000. Does the low number of patients with CF worldwide discourage scientists from finding a cure? Let’s have a look at what’s currently known about CF as well as existing and future therapies.

Characteristics of CF
CF is a highly underdiagnosed genetic disorder which is caused by a single defect gene. This gene is important for the development of a crucial protein, cystic fibrosis transmembrane conductance regulator (CFTR).
A defect CFTR gene is autosomal recessive, which means that an individual must inherit two faulty genes, one each from mother and father, to develop CF. People with one defect CFTR gene are called carriers. They develop no CF symptoms because the intact CFTR gene compensates for the faulty gene. If both parents are carriers of a faulty CFTR gene then the child will have a 50% chance of being a carrier, a 25% chance of developing CF and a 25% chance of being a healthy non-carrier.

A defect CFTR gene results in absent or faulty CFTR, which causes abnormalities in salt and fluid transport across epithelial cells. Epithelial cells are present in multiple organs, and CFTR helps to keep these organs moist by producing a liquid barrier.
Patients with CF develop many different symptoms, as the loss of CFTR affects different parts of the body:
- Sinuses and lungs lose their ability to maintain a functioning surface.
- The intestines get blocked by improperly hydrated stools.
- The pancreas and liver are unable to function normally and become permanently damaged. A poorly working pancreas is unable to absorb a lot of nutrients, which results in poor growth and no weight gain, despite excessive eating.
- The male reproductive tract is affected, with 98% of men being infertile.
The severity of the disease varies greatly among individuals with CF. However, deterioration is inevitable and eventually leads to death.

Effect of CF in the airways of lungs
The detailed mechanisms of CF development in the airways (respiratory system) remain unclear, as the function of CFTR and its interaction with other cell parts is complex. However, scientists have identified the following processes.
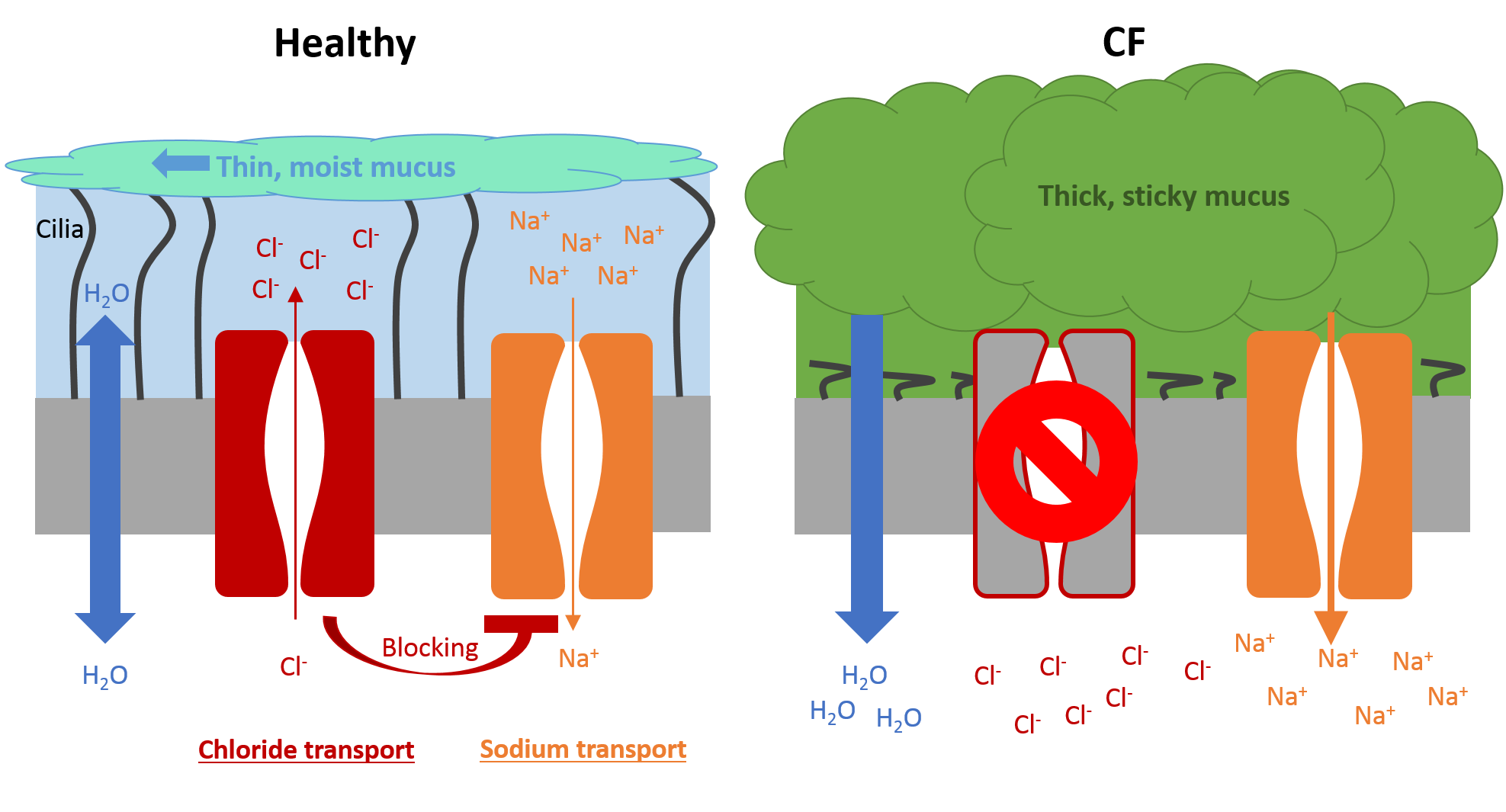
- In healthy individuals, the lungs are lined with epithelial cells that contain CFTR on the surface. CFTR enables hydration of the lung surface by transporting chloride ions (Cl–) and blocking sodium ion (Na+) channels. The increased accumulation of ions results in water movement, thereby creating a liquid layer on the lung surface. This layer allows cilia (tiny hairs) to transport any invading bacteria trapped in the mucus towards the mouth by coordinated ciliary beating. This clearance mechanism cleans the airways, with coughing if required. Apart from chloride and sodium ions, the liquid layer also includes many proteins and molecules that protect the lungs by inactivating and destroying invading bacteria and other pathogens.
- In individuals with CF, the lack of CFTR results in a thick and sticky mucus which flattens the cilia, thereby reducing the removal of invading bacteria. The thick mucus also blocks small airways and provides a perfect environment for bacteria to live in and cause infections. The immune system tries to eliminate the bacteria with an extensive pro-inflammatory response. An individual with CF has ten times more inflammation in their airways compared with a person without CF. The exact mechanisms of this inflammation are still unclear. Over time, the increased inflammation damages the airways, and most individuals with CF die of lung diseases.
The different types of CF
Scientists have discovered more than 1500 different mutations within the CFTR gene, and the consequences of many of these mutations are poorly understood. Each mutation can have varying effects on the function of CFTR; with some mutations resulting in milder forms of CF. Scientists have been able to group these mutations into six different classes according to CFTR status:
- Class 1: No CFTR is produced.
- Class 2: CFTR is produced, but incorrectly.
- Class 3: CFTR is correctly produced and is in the right place, but does not function as it should.
- Class 4: CFTR does not function properly as ions are unable to move through the channel.
- Class 5: CFTR is produced, but in very small quantities.
- Class 6: CFTR is produced correctly, but is dismantled too quickly by cellular mechanisms.
Therapies for CF
Most patients with CF receive help from medical teams in specialist centres. There is currently no cure. Historically, therapies only treated the symptoms of CF (mucus, lung infection and inflammation) in order to improve patients’ quality of life. These therapies include:
- Nutritional supplements and pancreatic enzymes to compensate for pancreatic malfunction
- Clearance of mucus from the lungs to reduce lung inflammation and improve lung function by using the following therapies:
- Physiotherapy, including postural drainage and chest clapping
- Antibiotics to clear bacterial infections
- Hypertonic saline to increase hydration of the airway surface
- An enzyme (deoxyribonuclease; also called DNase) to reduce the stickiness of the mucus
- For individuals with severe CF, transplantation is the only choice. These patients often receive multiple organ transplants of lung, heart and liver. However, patients receiving transplants are required to take immunosuppressive drugs, which are associated with substantial side effects.
More recently, therapies have become available which treat the causes of CF at the cellular level. However, due to the high number of possible mutations in the CFTR gene, these specific treatments are currently not able to help all patients. Therefore, further research is needed to treat more patients. A selection of cell-specific treatments at different stages of drug development is shown below.
- Read-through agents, which allow production of CFTR by ignoring any early termination signals that usually stop this process.
- Correctors, which improve the processing of CFTR and correct any faulty production.
- Potentiators, which increase the function of CFTR.
- Stimulation of alternative chloride channels to compensate for the lack of CFTR.
- Blockage of sodium absorption through blocking of sodium channels.
In addition, different types of gene therapies are in development, and a few studies have shown promising results in patients with CF. However, current methods do not produce enough working CFTR to fully cure the condition. Therefore, more intensive gene therapies that involve either dose increase or more frequent dosing are being carefully tested. However, these approaches are not without challenges: higher doses can cause more side effects and repeated dosing can activate the immune system, thereby weakening any therapeutic effects.
CF – the ongoing challenge
Despite advances in therapies within the past few years, patients with CF still have a reduced life expectancy. Thankfully, scientists have taken on the challenge of finding a cure for CF. They are working hard to develop new therapies, a few of which have shown encouraging results and may become available for a larger number of patients with CF in the future. However, further research is needed to completely understand the mechanisms involved in CF and to ultimately find a cure for this devastating disease.
Susanne Ulm is a Medical Writer part of our Prime team at Prime Global and has been with the company since November 2014. Susanne has broad experience in different therapeutic areas, and has a passion for communicating science to different types of audiences.


